







































































































 免费参与·100+跨境活动
免费参与·100+跨境活动 
 免费下载·4000+跨境资料
免费下载·4000+跨境资料 
 免费学习·2000+直播课程
免费学习·2000+直播课程 
 免费加入·15万+卖家交流群
免费加入·15万+卖家交流群 


2020-04-02 09:17

呼吸机是一种能够起到预防和治疗呼吸衰竭,减少并发症,挽救及延长病人生命的至关重要的医疗设备。
01出口通关要求
商品归类
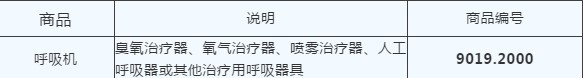
禁限管理根据商务部 海关总署 国家药品监督管理局2020年第5号公告,须提供书面或电子声明,承诺出口产品已取得我国医疗器械产品注册证书,符合进口国(地区)的质量标准要求。
目前具有呼吸机出口资质的企业名单如下:
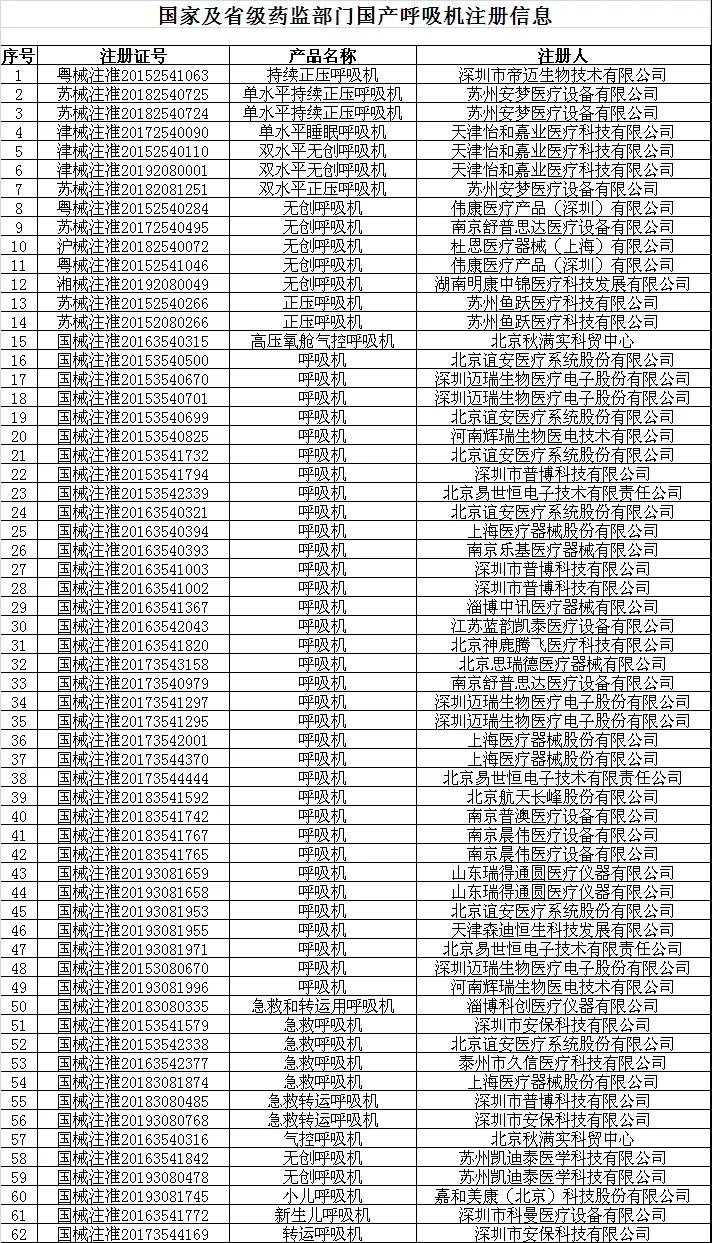
*该名单在国家药监局网站动态更新。
国家药监局网站动态更新查询地址:
http://www.nmpa.gov.cn/WS04/CL2582/

退税管理呼吸机及其配件的出口退税率为13%。
↓↓↓以下内容是根据国内外相关政府机构、↓↓↓
专业网站、新闻报道收集整理而成,
仅供参考。具体内容以相关管理部门、
国外官方机构要求为准。
相关货物出口时,中国海关无下列相关证件要求。
02各国呼吸机准入条件美国
必须要取得美国食品和药物管理局FDA注册认证才可以在美国本土市场进行销售,且FDA注册认证必须在美国本土办理,建议企业申请时委托美国本地代理。美国宣布自3月11日起取消部分医疗物资进口关税,其中包括呼吸机。中美关税查询地址:https://www.guanwuxiaoer.com/taxes.php
美国FDA对医疗器械的管理是通过器械与放射健康中心(CDRH)进行的,其对每一种医疗器械的分类和管理要求极其严格。
根据风险等级的不同,FDA将医疗器械分为三类(Ⅰ,Ⅱ,Ⅲ),呼吸机属于Ⅲ类高风险等级医疗设备。
在美国上市的呼吸机,须按照企业注册、产品列名、一般控制、特殊控制,向FDA递交510(k)等步骤逐步提交全部技术和临床文件,以期认证呼吸机的安全性、可靠性以及各项参数、性能等诸多方面,最终通过严谨的论证和试验来证明申请认证的机型可以完全满足FDA 的要求。
FDA 510(k)为医疗器械在美国上市的主要途径之一,绝大多数的II类医疗器械和部分I类、III类医疗器械通过此途径清关上市。
510(k)文件是向FDA递交的上市前申请文件,目的是证明所申请上市的产品和已在美国市场上合法销售的产品在安全性和有效性方面比较是实质相等的,即为等价器械(substantially equivalent)。
申请者必须把申请上市的器械与现在美国市场上一种或多种相似器械对比,得出并且支持等价器械的结论。对FDA 510(k)注册文件所必须包含的信息,FDA有一个基本的要求,其内容大致如下16个方面:
1.申请函此部分应包括申请人(或联系人)和企业的基本信息、FDA 510(K)递交的目的、申请上市器械的名称型号和分类资料、进行实质等效比较的产品(Predicate Device)名称及其510(k)号码。
2.目录即FDA 510(k)文件中所含全部资料的清单(包括附件)。
3.真实性保证声明FDA有一个标准的样本
4.器材名称即产品通用名、FDA分类名、产品贸易名。
5.注册号码如企业在递交FDA 510(k)时已进行企业注册,则应给出注册信息,若未注册,也予注明。
6.分类即产品的分类组、类别、管理号和产品代码。
7.性能标准产品所满足的强制性标准或自愿性标准。
8.产品标识包括企业包装标识、使用说明书、包装附件、产品标示等。
9.实质相等性比较(SE)选择合适的产品进行比较是510(K)申请的关键步骤。在进行比较时应从如下方面进行考虑:企业必须提供充足的资料证明,所申请上市的器械和被比较的器械是实质相等的(SE),否则510(k)申请不会通过。
10.510(k)摘要或声明申请文件摘要和支持等价器械的结论。
11.产品描述包括产品的预期用途、工作原理、动力来源、零组件、照片、工艺图、装配图、结构示意图等。
12.产品的安全性与有效性包括各种设计、测试资料。
13.生物相容性生物相容性是指材料与生物体之间相互作用后产生的各种生物、物理、化学等反应的一种概念。一般地讲,就是材料植入人体后与人体相容程度,也就是说是否会对人体组织造成毒害作用。
14.色素添加剂(如适用)
15.软件验证(如适用)
16.灭菌(如适用)包括灭菌方法的描述、灭菌验证产品包装和标识等。● 技术标准简析需特别注意的是,美标中要求婴儿呼吸机工作压力控制应在整个范围内精确至 ±2cm H2O,而其他呼吸机应精确至±5cm H2O。与美标相比,国标没有按照婴儿呼吸机和其他呼吸机进行要求,要求读数的精度为± (2%满刻度+4%实际读数)。另外就是国标对报警声音的要求是最新执行的标准,而美国的设备是按照声称的标准进行测试。
欧盟
须获得欧盟CE认证,并符合技术法规:MDD 93/42/EEC或MDR (EU) 2017/745。 建议找欧盟认可的公告机构进行检测。
1.欧盟官网MDD 93/42/EEC医疗器械指令授权的机构查询地址:
https://ec.europa.eu/growth/tools-databases/nando/index.cfm?fuseaction=directive.notifiedbody&dir_id=13
2.自2020年5月26日起,MDR (EU) 2017/745医疗器械法规将正式取代欧盟现行的MDD医疗器械指令强制实施,同样在欧盟官网可以查询到。
欧盟官网MDR (EU) 2017/745医疗器械法规授权的机构查询地址:
https://ec.europa.eu/growth/tools-databases/nando/index.cfm?fuseaction=directive.notifiedbody&dir_id=34
欧盟CE认证资格辨别可参见>欧盟CE认证资格辨别指南(附180家授权机构清单)
特别提醒:2020年5月26号施行的欧盟MDR对目前CE认证MDD而言有哪些影响?● 目前大部分CE证书是按照MDD要求测试的,面临1月后MDR的换证问题;
● 拥有MDD授权的公告机构,并不全是MDR授权的公告机构,CE认证审核机构可选性降低;
● MDR的审核要求比MDD更为复杂,认证周期必然大幅度拉长;
● CE认证费用可能将有大幅提升;
● 欧盟对医疗设备的监管更加严格。
日本
如果需要投放市场产品必须满足日本的Pharmaceutical and Medical Device Act (PMD Act),在PMD Act的要求下,TOROKU注册系统要求国外的制造商必须向PMDA注册制造商信息。
日本医药品和医疗器械综合机构(PMDA)
网址:www.pmda.go.jp
韩国
韩国医疗器械准入的法规门槛,基本分类为I、II、III、IV类,持证为韩国公司(License holder),韩国收货人需要到韩国药监局Korea Pharmaceutical Traders Association. 必须提前备案进口资质。韩国药监局Korea Pharmaceutical Traders Association网址:www.kpta.or.kr
澳大利亚
须通过澳洲的TGA注册,TGA 是Therapeutic Goods Administration的简写,全称是治疗商品管理局。澳大利亚对医疗器械分为I类,Is and Im, IIa, IIb, III类,分别为豁免、备案和注册。无论哪类医疗器械,其上市销售前必须得到澳大利亚政府的准许,符合医疗器械的基本要求,按照符合性审查程序进行审查。
特别提醒:澳大利亚已与欧盟达成互认协议。这意味着,合格评定证书由TGA颁发的也被欧盟认可,TGA也认可欧盟CE认证。已获CE认证的用户,可提交CE证书及相关资料,获得TGA证书。
如果产品已经注册或备案,制造商更换经销商对其没有影响。对国外产品进行注册审批后,每年还要常规注册一次,说明产品型号、性能及质量有无变化。TGA 全权负责对医疗器械的符合性评价,并收取一定费用,相关费用金额可参见 TGA的网站。
TGA网址:https://www.tga.gov.au/
03各国呼吸机技术标准简析
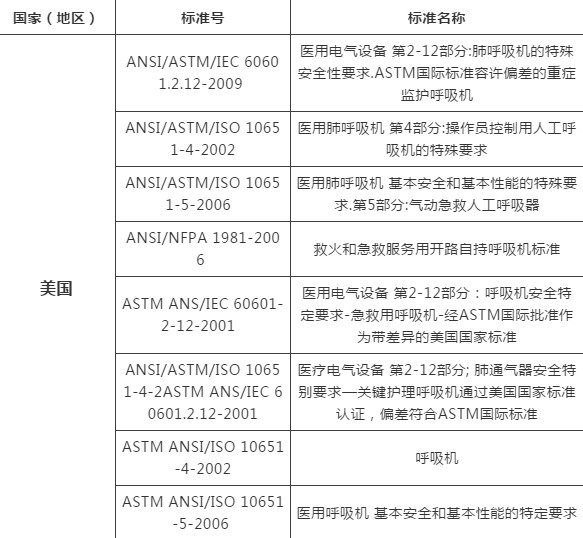
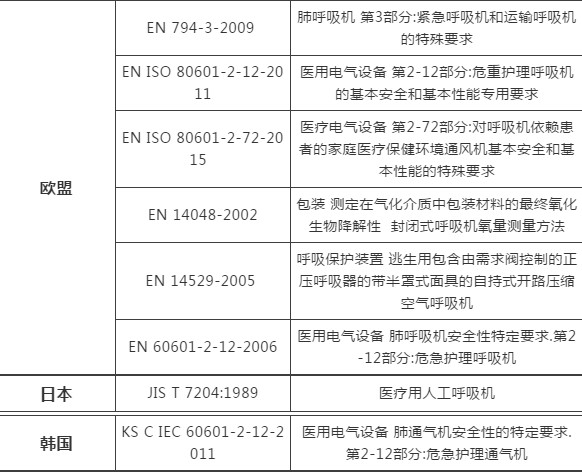
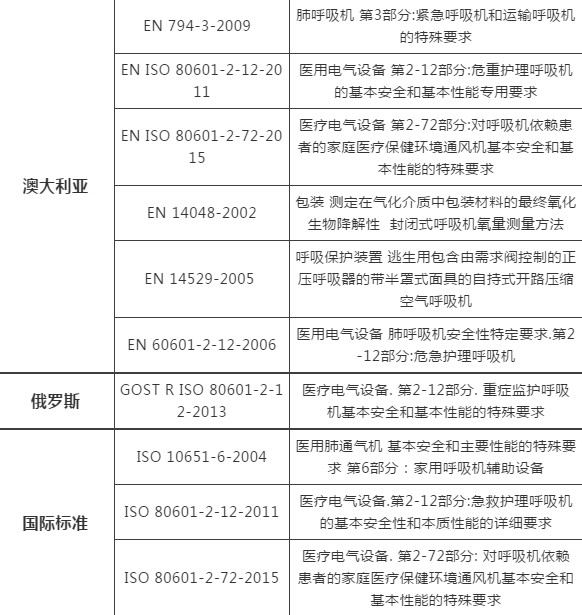
*以上技术标准为各国进口或销售时,由各国海关或相关部门要求验核;如有动态调整,以相关标准管理机构官方发布为准,仅作为提醒或参考资料。
(来源:关务小二)
以上内容属作者个人观点,不代表雨果网立场!如有侵权,请联系我们。

















 闽公网安备35020602003453号
闽公网安备35020602003453号