






















































































 免费参与·100+跨境活动
免费参与·100+跨境活动 
 免费下载·4000+跨境资料
免费下载·4000+跨境资料 
 免费学习·2000+直播课程
免费学习·2000+直播课程 
 免费加入·15万+卖家交流群
免费加入·15万+卖家交流群 


2020-03-24 10:38

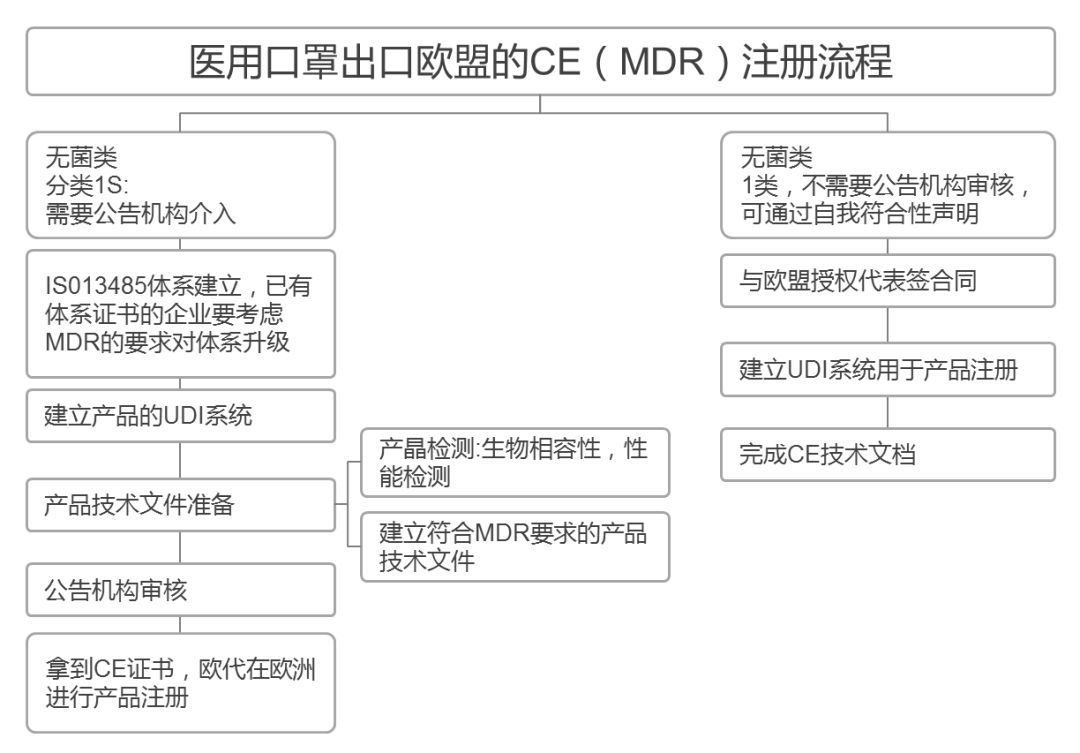
面对新冠疫情,为满足口罩、防护服等防疫用品日益增长的需求,欧盟开启绿色通道,无需CE标识,可进入欧盟,供医疗工作者使用。允许部分防疫物资(如一类灭菌的医用口罩)在符合安全有效的情况下,即使尚未获得CE认证,也可以在欧盟市场上市销售。
3月17日,美国FDA对新冠病毒检测产品也开通了类似的上市绿色通道,允许检测试剂盒在还未获EUA授权之前,可以先进入临床市场。目前,包括华大基因在内的多家国内企业均已启动产品在美国临床市场的商业销售。
2020年3月13日,欧盟会员会在欧洲官方杂志( Official Journal of the EuropeanUnion)发布了疫情期间针对医疗器械和个人防护用品 (PPE)的符合性评价和市场监督程序的建议。
医疗器械方面:

◆如果市场监督机构确定产品符合医疗器械的基本安全和性能要求,即使其符合性评价还未完成,市场监督机构可以允许其在一定的时间内进行销售,同时该产品必须继续完成其符合性评价过程。
◆成员国主管当局也可在疫情期间评估和组织采购没有CE标记的医疗器械,该产品仅可提供给医疗工作者使用,不能在市场上流通销售。同时市场抽查将会重点抽查防疫相关医疗器械,以防止不合格产品导致严重风险。
个人防护用品(PPE)方面:

◆涉及的产品包括抛弃式和可重复使用的口罩、可重复使用的工作服、手套和眼罩等(主要是预防病毒和有害物质的产品)。需要具有PPE法规授权资格的公告机构进行符合性评价。
◆应急审批产品如果不采用PPE法规协调标准作为产品技术要求而采用其它技术要求,比如WHO的推荐要求,须确保采用的技术要求与PPE法规基本健康与安全要求同等防护水平。公告机构对这类采用其它技术要求的PPE产品进行发证时,需要立即通知主管当局和其它PPE法规的公告机构。
◆如果市场监督机构确定产品符合PPE法规的基本健康和安全要求,即使其符合性评价还未完成,市场监督机构可以允许其在一定的时间内进行销售,同时该产品必须继续完成其符合性评价过程。
◆成员国主管当局也可在疫情期间评估和组织采购没有CE标记的PPE产品,该产品仅可提供给医疗工作者使用,不能在市场上流通销售。同时市场抽查将会重点抽查防疫相关PPE产品,以防止不合格产品导致严重风险。
也就是说,只要处于正在进行符合性评估的过程中,就可以在没有CE标志的情况下先行进入欧盟市场。由市场监督部门进行抽查,发现问题再进行处罚。
重点如下!!!

◆成员国可采购安全有效,但没有CE标记的医疗产品;
◆紧急物资专供医疗人员使用,不可在市场上流通;
◆仅疫情期间有效。
关于CE标识

CE标签就像一把巨伞,底下是规定各类产品安全标准、细分到不同材料和生产模式等的各种欧盟指令。自1985年成立以来,它就成为了高质量、高标准和严格执法的标志,缺少这一标志的商品将不予获准进入欧盟市场。
如今CE标识已经成为了全球认可的质量标志,CE标志可以证明该批在欧盟制作或进口至欧盟成员国的产品符合质量标准,满足保护消费者健康、供应链安全和环境可持续发展的要求。
在欧盟,口罩属于PPE个人防护用品,“危及健康的物质和混合物”。2019年起,欧盟新法规PPE Regulation (EU) 2016/425强制执行,所有出口欧盟的口罩必须在新法规的要求下获得CE认证证书。CE认证证书的有效期是5年左右,一般费用是10000-15000元人民币。
欧盟对于口罩欧洲统一,CE认证的标准包括BSEN140、BSEN14387、BSEN143、BSEN149、BSEN136,其中BSEN149使用多,为可防护微粒的过滤式半口罩,根据测试的粒子穿透率分为P1(FFP1),P2(FFP2),P3(FFP3)三个等级,FFP1低过滤效果≥80%,FFP2低过滤效果≥94%,FFP3低过滤效果≥97%。
FFP2口罩与上文提到的医用防护口罩、KN95口罩、N95口罩过滤效率十分接近。医疗口罩必须遵循BSEN14683标准,可以分为三个等级:低标准Type、然后是Type和TypeR。上一个版本是BSEN146832014,已被新版BSEN146832019所取代。EN 14683:2019年版主要的变化之一是压力差,Type、Type、TypeR压力差分别由2014年版的29.4、29.4、49.0Pa/cm,上升至40、40、60Pa/cm。
CE认证是欧盟实行的强制性产品安全认证制度,目的是为了保障欧盟国家人民的生命财产安全。

新手小白可以问自己的发证机构两个问题:
01、贵司是否为NB机构? 机构号是否可以查询?
02、出具的CE证书在官网可查吗?

NB机构可以理解为被欧盟授权或认可的机构。如果CE证书是NB机构发证的,在欧盟就具有一定的效应,清关的风险才会相对较小。
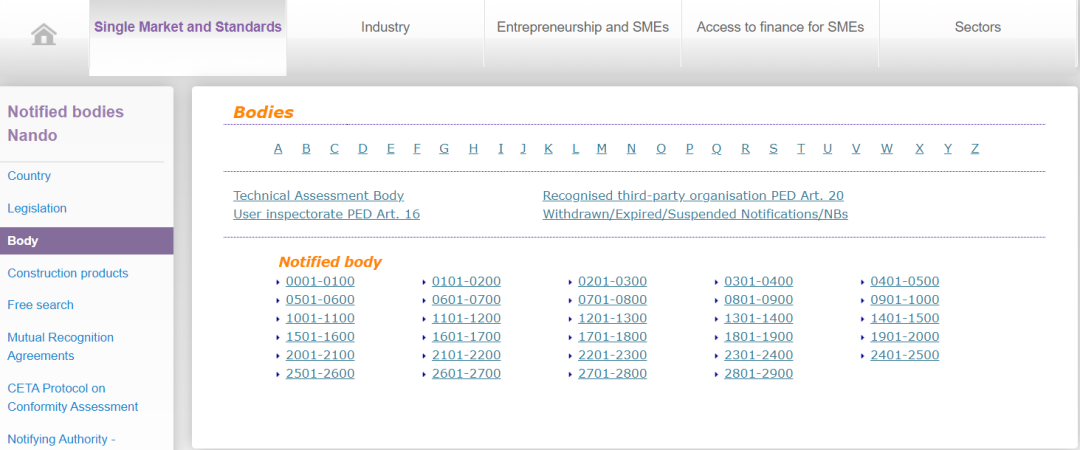
正常情况下,根据欧盟法规,所有出口欧盟的产品都需要获得CE认证,加贴CE标识才能进入欧洲市场。CE认证的审核和发证,欧盟公布了一系列由欧盟统一监管和认证资质授权的机构,并授予每家机构一个唯一的四位数编码即公告号,CE证书的申请和颁发就由对应法规和指令授权的公告号机构颁发。
在欧盟官方网站-欧盟公告机构查询官网,厂家可以查询到目前从0001-2786 两千多家欧盟公告号机构详细信息,每家机构对应的指令和法规授权以及发证机构信息都可在该网站查询到。
附:CE认证查验:
https://ec.europa.eu/growth/tools-databases/nando/index.cfm?fuseaction=notifiedbody.main
附:FDA查验(出口美国需要FDA和NIOSH):
https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfRL/rl.cfm
近期,大家都在对口罩等医疗物资的欧盟标准犯愁,都是一知半解的状态,今天Mike将欧盟通告原文贴出,大家可以自己好好研究一下每种标准的具体陈述;昨天给了链接后,很多人说打不开网页,其实应该是你们的局域网问题,不过没关系,我给你们贴出来:
COMMISSION RECOMMENDATION (EU) 2020/403
of 13 March 2020
on conformity assessment and market surveillance procedures within the context of the COVID-19 threat
THE EUROPEAN COMMISSION,
Having regard to the Treaty on the Functioning of the European Union, and in particular Article 292 thereof,
Whereas:
01
In the context of the current COVID-19 global outbreak as well as the rapid spread of the virus across various regions of the EU, the demand for personal protective equipment (hereinafter ‘PPE’) such as face masks, gloves, protective coveralls or eyewear protection, as well as for medical devices such as surgical masks, exploration gloves and some gowns, has seen an exponential growth. In particular, the supply chain of certain types of PPE such as the disposable face masks is under severe strain, due to the exponential growth of the demand both via existing as well as via new channels. In addition, the global supply chain of such products has also sustained significant disruptions, which have induced repercussions on the EU market as well.
02
Bearing in mind that the health and safety of the EU citizens is of upmost priority, it is of paramount importance to ensure that the most appropriate PPE and medical devices ensuring adequate protection are swiftly made available to those who need it most.
03
Economic operators active across the EU are working relentlessly to increase their respective manufacturing and distribution capacity. In order to mitigate the effects of the various disruptive factors, the economic operators are redesigning their supply chains by launching new manufacturing lines and/or diversifying their supplier base. These efforts by the industrial stakeholders would not be able to produce their full effects if the increased supply cannot feed into the market without any undue delays.
04
The requirements for the design, manufacturing and placing on the market of personal protective equipment are laid down by Regulation (EU) 2016/425 of the European Parliament and of the Council of 9 March 2016 on personal protective equipment and repealing Council Directive 89/686/EEC (1).
05
The requirements for the design, manufacturing and placing on the market of medical devices are laid down by Council Directive 93/42/EEC of 14 June 1993 concerning medical devices (2). That Directive is repealed by Regulation (EU) 2017/745 of the European Parliament and of the Council of 5 April 2017 on medical devices, amending Directive 2001/83/EC, Regulation (EC) No 178/2002 and Regulation (EC) No 1223/2009 and repealing Council Directives 90/385/EEC and 93/42/EEC (3), with effect from 26 May 2020.
06
Disposable and re-usable face masks ensuring protection against particulate hazards, disposable and re-usable coveralls, gloves and eyewear protection, which are used for prevention and protection against harmful biological agents such as viruses are products falling within the scope of the Regulation (EU) 2016/425.
07
Surgical masks, examination gloves and some types of gowns are products falling within the scope of Directive 93/42/EEC and of Regulation (EU) 2017/745.
08
In the context of the COVID-19 threat, such PPE and medical devices are essential for healthcare workers, first responders and other personnel involved in the efforts to contain the virus and avoid its further spread.
09
Regulation (EU) 2016/425 fully harmonises the rules for the design, manufacturing and placing on the Union market of PPE and sets out a number of essential health and safety requirements for PPE based on a classification of PPE depending on the risk against which it is intended to protect users. Thus, items of PPE manufactured in accordance with the Regulation (EU) 2016/425 can circulate freely throughout the internal market and Member States may not introduce additional and diverging requirements regarding the manufacturing and placement on the market of such products.
10
Directive 93/42/EEC and Regulation (EU) 2017/745 fully harmonise the rules for the design, manufacturing and placing the Union market of medical devices, and set up a number of essential requirements and of general safety and performance requirements, based on a classification of medical devices depending on specific rules governed by the intended purpose of the devices. Thus, devices manufactured in accordance with the Council Directive 93/42/EEC and Regulation (EU) 2017/745 can circulate freely throughout the internal market and Member States may not introduce additional and diverging requirements regarding the manufacturing and placement on the market of such products.
11
PPE intended to protect against harmful biological agents, such as viruses are listed in Annex I of Regulation (EU) 2016/425 as category III, which includes exclusively the risks that may cause ‘very serious consequences such as death or irreversible damage to health’.
12
Relevant medical devices as non-invasive devices are in Class I, unless specific rules apply.
13
In accordance with Article 8 of Regulation (EU) 2016/425, in order to place PPE products on the market, manufacturers shall carry out the applicable conformity assessment procedures and, where compliance with the applicable essential health and safety requirements has been demonstrated by the appropriate procedure, affix the CE marking.
14
In accordance with Article 11 of Directive 93/42/EEC and with Article 52 of Regulation (EU) 2017/745, once the latter becomes applicable, in order to place medical devices on the market, manufacturers shall carry out the applicable conformity assessment procedures and, where compliance with the applicable essential requirements or general safety and performance requirements has been demonstrated by the appropriate procedure, affix the CE marking. Derogations from conformity assessment procedures may be authorised by Member States, on duly justified request, for the placing on the market and putting into service, within the territory of the Member State concerned, of individual devices the use of which is in the interest of protection of health.
15
Regulation (EU) 2016/425 is technologically neutral and does not lay down any specific mandatory technical solutions for the design of PPE products. Instead, Annex II to Regulation (EU) 2016/425 sets the essential health and safety requirements, which PPE should meet in order to be able to be placed on the market and to circulate freely across the entire EU market.
16
Directive 93/42/EEC and Regulation (EU) 2017/745 are technologically neutral and do not lay down any specific mandatory technical solutions for the design of medical devices. Instead, Annex I to Directive 93/42/EEC sets the essential requirements, and Annex I to Regulation (EU) 2017/745 sets the general safety and performance requirements, which medical devices should meet in order to be able to be placed on the market and to circulate freely across the entire EU market.
17
Article 14 of Regulation (EU) 2016/425 offers the possibility for manufacturers to rely on specific technical solutions, which are detailed in harmonised standards or parts thereof the references of which have been published in the Official Journal of the European Union. In accordance with this Article, should a manufacturer choose to adopt such a technical solution, the PPE is presumed to be in conformity with the essential health and safety requirements covered by the said harmonised standard or parts thereof. However, compliance with the harmonised standards is not mandatory. Manufacturers are free to choose other technical solutions provided that the specific solution which is retained ensures that the PPE complies with the applicable essential health and safety requirements.
18
Article 5 of Directive 93/42/EEC and Article 8 of Regulation (EU) 2017/745 offer the possibility for manufacturers to rely on specific technical solutions, which are detailed in harmonised standards or parts thereof the references of which have been published in the Official Journal of the European Union. In accordance with this Article, should a manufacturer choose to adopt such a technical solution, the medical device is presumed to be in conformity with the requirements covered by the said harmonised standard or parts thereof. However, compliance with the harmonised standards is not mandatory. Manufacturers are free to choose other technical solutions provided that the specific solution which is retained ensures that the medical device complies with the applicable essential health and safety requirements.
19
Article 19 of Regulation (EU) 2016/425 lays down the specific conformity assessment procedures, which apply to the different categories of PPE. Pursuant to this Article, items of PPE of category III, such as the ones designed protect against harmful biological agents should be subjected to specific combination of conformity assessment procedures, which are described respectively in Annexes V, VII and VIII of the same Regulation. Each of the different conformity assessment procedures, which may be used, require the mandatory involvement of a third party conformity assessment body.
20
Article 11 of Directive 93/42/EEC and Article 52 of Regulation (EU) 2017/745, once the latter becomes applicable, lay down the specific conformity assessment procedures, which apply to the different classes of medical devices. Pursuant to these Articles, medical devices falling within Class I, other than custom-made or investigational devices, should be subjected to the conformity assessment procedure for the EC declaration of conformity, without the involvement of a third party conformity assessment body.
21
Notified bodies are the conformity assessment bodies designated by Member States and authorised to carry out third party conformity assessment tasks under Regulation (EU) 2016/425. According to Article 26(4) and point 7 (f) of Annex V of Regulation (EU) 2016/425, notified bodies are required to assess that a PPE product meets the applicable essential health and safety requirements. Notified bodies need to carry out this assessment not only where the manufacturer has applied the harmonised standards, but also in a situation where the manufacturer has followed other technical solutions. When delivering the conformity assessment certificates, notified bodies are required to inform their notifying authorities and may also be required to inform other notified bodies of the certificates they have issued, as set out in Article 34 of Regulation (EU) 2016/425.
22
Notified bodies should thus assess whether products manufactured in line with other technical solutions, such as the ones contained in the WHO recommendations on the appropriate selection of PPE also meet the applicable essential health and safety requirements. In view of the importance to ensure an efficient exchange of information between all stakeholders in the PPE supply chain, where notified bodies conclude that a PPE following another specific standard or technical solution is compliant with the essential health and safety requirements applicable to it, sharing this information will be instrumental in facilitating the assessment of other products manufactured according to the same specific standard or technical solution in a swift manner. To that effect, notified bodies can make use of the existing channels for exchange of information in the framework of the coordination groups established in accordance with Article 36 of Regulation (EU) 2016/425.
23
In addition, pursuant to the relevant market surveillance procedures in Regulation (EU) 2016/425 and in particular Article 38(1) and (2) thereof, where a market surveillance authority encounters a non-CE marked PPE product they are required to evaluate it. Where, in the course of the evaluation, the market surveillance authorities find that the PPE does not comply with the requirements laid down in the Regulation, they shall require the economic operator to take corrective action to bring the PPE into compliance or to recall or withdraw it, commensurate with the nature of the risk. They shall also inform the Commission and other Member State of the results of the evaluation and the actions which they have required the economic operator to take.
24
Accordingly, to address the shortage of PPE necessary in the context of the COVID-19 outbreak, where non-CE marked PPE are intended to enter the EU market, the relevant market surveillance authorities should evaluate the products and, if they are found to be compliant with the essential health and safety requirements laid down by the relevant Regulation should take measures allowing the placing of such PPE on the Union market for a limited period of time or while the conformity assessment procedure with the notified body is being carried out. In order to ensure that such products can be made available in other Member States and in view of the importance to ensure an efficient exchange of information as well as a coordinated response to all threats to the citizens’ health and safety, it is appropriate that the market surveillance authority carrying out such an evaluation communicates its decision to other Member States authorities and to the Commission through the regular market surveillance information exchange channels.
25
Considering that certain types of PPE or medical devices that are used in the context of the COVID-19 outbreak, may also be used for other purposes, it is necessary that Member States take all appropriate measures to ensure that PPE or medical devices not bearing the CE marking, which may be placed on the Union market in accordance with paragraph 8 of the present Recommendation are only made available to healthcare workers,
HAS ADOPTED THIS RECOMMENDATION:
1. With the objective to ensure availability of PPE and medical devices for adequate protection in the COVID-19 outbreak, the Commission invites all economic operators throughout the supply chain, as well as notified bodies and market surveillance authorities to deploy all the measures at their disposal to support the efforts aimed at ensuring that the supply of PPE and medical devices throughout the EU market will match the continuously increasing demand. Such measures should nevertheless not have a detrimental effect on the overall level of health and safety and all relevant stakeholders should ensure that any PPE or medical devices, which is being placed on the EU market, continues to provide an adequate level of protection of the users’ health and safety.
CONFORMITY ASSESSMENT PROCEDURES
2.The notified bodies under Regulation (EU) 2016/425 should prioritise and swiftly conduct the conformity assessment activities in the framework of all newly submitted requests by economic operators of PPE necessary for protection in the context of the COVID-19 outbreak.
3.In the case of PPE products manufactured following technical solutions other than harmonised standards, the WHO recommendations on the appropriate selection of PPE may be used as a potential source of reference for such technical solutions, provided that the said technical solutions ensure an adequate level of protection corresponding to the applicable essential health and safety requirements laid down in Regulation (EU) 2016/425.
4.Notified bodies which issue certificates to PPE products manufactured following other technical solutions than harmonised standards, should immediately inform the relevant notifying authority as well as the other notified bodies under Regulation (EU) 2016/425 of the certificates issued and the specific technical solution followed. Notified bodies should exchange such information through the coordination of notified bodies group established under Article 36 of Regulation (EU) 2016/425.
5.In the case of medical devices, the possibility for Member States to authorise derogations from conformity assessment procedures should also be considered, according to Article 11(13) of Directive 93/42/EEC and Article 59 of Regulation (EU) 2017/745 once the latter becomes applicable, also when the intervention of a notified body is not required.
MARKET SURVEILLANCE PROCEDURES
6.The relevant market surveillance authorities in the Member States should as a matter of priority focus on non-compliant PPE or medical devices raising serious risks as to the health and safety of their intended users.
7.Where market surveillance authorities find that PPE or medical devices ensure an adequate level of health and safety in accordance with the essential requirements laid down in Regulation (EU) 2016/425 or the requirements of Directive 93/42/EEC or Regulation (EU) 2017/745, even though the conformity assessment procedures, including the affixing of CE marking have not been fully finalised according to the harmonised rules, they may authorise the making available of these products on the Union market for a limited period of time and while the necessary procedures are being carried out.
8.PPE or medical devices not bearing the CE marking could also be assessed and part of a purchase organised by the relevant Member State authorities provided that is ensured that such products are only available for the healthcare workers for the duration of the current health crisis and that they are not entering the regular distribution channels and made available to other users.
9.Market surveillance authorities should inform immediately the Commission and other Member States of any temporary arrangement they have granted to specific PPE or medical devices. For PPE, this should be done through the Information and Communication System for Market Surveillance (ICSMS).
Done at Brussels, 13 March 2020.
For the Commission
Thierry BRETON
Member of the Commission
原文查看(实在打不开的,建议换浏览器或电脑试试): https://eur-lex.europa.eu/legal-content/EN/TXT/HTML/?uri=CELEX:32020H0403&from=EN
COVID-19威胁范围内的合格评定和市场监督程序
(翻译版供参考,具体请以英文原版为准)
欧洲委员会,考虑到《欧洲联盟运作条约》,特别是其第292条,鉴于:
1、在当前的COVID-19全球爆发以及病毒在欧盟各个地区的快速传播的背景下,对个人防护设备(以下称“ PPE”)的需求,例如口罩,手套,防护工作服或眼镜防护以及用于外科口罩,探查手套和某些礼服等医疗设备的数量呈指数增长。特别是,由于通过现有渠道以及通过新渠道的需求呈指数增长,某些类型的PPE(例如一次性口罩)的供应链承受着巨大的压力。此外,此类产品的全球供应链也遭受了严重破坏,这也引起了欧盟市场的反响。
2、牢记欧盟公民的健康和安全是重中之重,因此,确保向最需要的人迅速提供最适当的个人防护设备和医疗设备,以确保提供足够的保护,这一点至关重要。
3、活跃于整个欧盟的经济运营商正在不懈努力,以提高各自的制造和分销能力。为了减轻各种破坏性因素的影响,经济运营商正在通过启动新的生产线和/或使其供应商基础多样化来重新设计其供应链。如果供应增加而没有任何不适当的延误,则工业利益相关者的这些努力将无法发挥全部作用。
4、有关个人防护设备的设计,制造和投放市场的要求,由欧洲议会和理事会于2016年3月9日颁布的第(EU)2016/425号条例(关于个人防护设备)以及废除理事会第89/686号指令规定/ EEC (1)。
5、1993年6月14日关于医疗器械的理事会指令93/42 / EEC对医疗器械的设计,制造和投放市场提出了要求 (2)。欧洲议会和2017年4月5日关于医疗器械的理事会(EU)2017/745条例,该指令2001/83 / EC,条例(EC)178/2002和条例(EC)的修订已废除该指令1223/2009和废除理事会指令90/385 / EEC和93/42 / EEC (3),自2020年5月26日起生效。
6、一次性和可重复使用的口罩,可防止颗粒物危害,一次性和可重复使用的工作服,手套和眼镜防护,用于预防和防止有害生物制剂(如病毒)属于本法规范围内的产品(欧盟)2016/425。
7、外科口罩,检查手套和某些长袍类型的产品属于93/42 / EEC指令和2017/745法规(EU)的范围。
8、在发生COVID-19威胁的情况下,此类PPE和医疗设备对于参与遏制病毒并避免其进一步传播的医护人员,急救人员和其他人员至关重要。
9、(EU)2016/425法规充分协调了PPE的设计,制造和投放市场的规则,并根据PPE的类别(取决于其所面临的风险)对PPE提出了一些基本的健康和安全要求旨在保护用户。因此,根据法规(EU)2016/425制造的PPE物品可以在整个内部市场自由流通,成员国不得对此类产品的制造和投放市场引入额外的和不同的要求。
10、93/42 / EEC指令和(EU)2017/745法规充分协调了医疗器械联盟市场的设计,制造和销售规则,并根据以下内容建立了一些基本要求以及一般安全和性能要求根据受器械预期用途支配的特定规则对医疗器械进行分类。因此,根据理事会指令93/42 / EEC和法规(EU)2017/745制造的设备可以在整个内部市场自由流通,成员国不得对此类设备的制造和投放市场引入额外的和不同的要求产品。
11、PPE旨在防止有害生物制剂,如病毒在法规附件I(EU)四百二十五分之二千零十六为III类,其中包括完全可能导致的风险上市“非常严重的后果,如死亡或不可逆的损害健康 ” 。
12、除非适用特定规则,否则与非侵入性设备相关的医疗设备属于I类。
13、根据(EU)2016/425号法规第8条,为了将PPE产品投放市场,制造商应执行适用的合格评定程序,并在符合条件的情况下证明符合适用的基本健康和安全要求按照适当的程序,贴上CE标志。
14、根据指令93/42 / EEC的第11条和(EU)2017/745法规的第52条,一旦后者适用,为了将医疗设备投放市场,制造商应执行适用的合格评定程序并且,如果通过适当的程序证明符合适用的基本要求或一般安全和性能要求,请贴上CE标志。会员国可应正当理由要求批准从合格评定程序中减损,以便在有关会员国领土内投放市场并在使用中保护受保护的单个设备健康。
15、法规(EU)2016/425在技术上是中立的,没有为PPE产品的设计规定任何特定的强制性技术解决方案。相反,法规(EU)2016/425的附件II规定了基本的健康和安全要求,PPE应该满足这些基本要求,以便能够投放市场并在整个欧盟市场上自由流通。
16、93/42 / EEC指令和法规(EU)2017/745在技术上是中立的,没有为医疗设备的设计规定任何特定的强制性技术解决方案。相反,指令93/42 / EEC的附件I设置了基本要求,法规(EU)2017/745的附件I设置了一般安全性和性能要求,医疗设备应满足这些要求才能将其放置在市场,并在整个欧盟市场上自由流通。
17、(EU)2016/425号法规第14条为制造商提供了依赖特定技术解决方案的可能性,这些解决方案在统一标准或其部分中进行了详细说明,其参考文献已在《欧盟官方杂志》上发表。根据本条,如果制造商选择采用这样的技术解决方案,则假定个人防护设备符合上述统一标准或其部分所涵盖的基本健康和安全要求。但是,并非必须遵守协调标准。制造商可以自由选择其他技术解决方案,前提是保留的特定解决方案可确保PPE符合适用的基本健康和安全要求。
18、指令93/42 / EEC的第5条和法规(EU)2017/745的第8条为制造商提供了依赖特定技术解决方案的可能性,这些解决方案在统一标准或其部分中作了详细说明,其参考文献已在欧盟官方杂志。根据本条,如果制造商选择采用这种技术解决方案,则假定医疗器械符合上述协调标准或其部分所涵盖的要求。但是,并非必须遵守协调标准。制造商可以自由选择其他技术解决方案,前提是保留的特定解决方案可确保医疗设备符合适用的基本健康和安全要求。
19、(EU)2016/425号法规第19条规定了具体的合格评定程序,该程序适用于不同类别的PPE。根据本条,第三类个人防护装备的项目,例如旨在防止有害生物制剂的项目,应进行合格评定程序的特定组合,分别在同一法规的附件V,VII和VIII中进行描述。可能使用的每种不同的合格评定程序都需要第三方合格评定机构的强制参与。
20、93/42 / EEC指令的第11条和法规(EU)2017/745的第52条一旦适用,就制定适用于不同类别医疗设备的特定合格评定程序。根据这些条款,属于第一类的医疗设备(定制或研究用设备除外)应接受EC合格声明的合格评定程序,而无需第三方合格评定机构的参与。
21、公告机构是成员国指定的合格评定机构,并有权执行法规(EU)2016/425中的第三方合格评定任务。根据第(EU)2016/425号法规第26条第4款和附件V第7点(f)的规定,指定机构必须评估PPE产品是否符合适用的基本健康和安全要求。认证机构不仅需要在制造商采用统一标准的地方进行评估,还需要在制造商遵循其他技术解决方案的情况下进行评估。交付合格评定证书时,指定机构必须通知其通知当局,也可能需要将其签发的证书通知其他指定机构,
22、因此,公告机构应评估根据其他技术解决方案生产的产品(例如,世卫组织关于适当选择个人防护装备的建议中所包含的产品)是否也符合适用的基本健康和安全要求。考虑到确保个人防护装备供应链中所有利益相关者之间有效信息交换的重要性,公告机构得出结论认为,遵循另一项特定标准或技术解决方案的个人防护装备符合适用于其的基本健康和安全要求,这些信息将有助于快速评估根据同一特定标准或技术解决方案生产的其他产品。为此,
23、此外,根据法规(EU)2016/425中的相关市场监督程序,尤其是其中的第38(1)和(2)条,如果市场监督机构遇到非CE标志的PPE产品,则需要对其进行评估它。市场评估机构在评估过程中发现个人防护设备不符合本规章的要求时,应要求经济经营者采取纠正措施以使个人防护设备符合要求或召回或撤回它,与风险的性质相称。他们还应将评估结果以及他们要求经济经营者采取的行动告知委员会和其他成员国。
24、因此,为了解决在COVID-19爆发时(非CE标志的PPE打算进入欧盟市场)所必需的PPE短缺的问题,相关的市场监督机构应对产品进行评估,如果发现这些产品属于符合相关法规所规定的基本健康与安全要求的人员,应采取措施,在有限的时间内或在与指定机构进行合格评定程序的同时,将此类PPE投放到欧盟市场。为了确保可以在其他会员国提供此类产品,并考虑到确保有效交换信息以及对对公民健康和安全的所有威胁作出协调反应的重要性,
25、考虑到在COVID-19爆发中使用的某些类型的PPE或医疗设备也可能用于其他目的,有必要使会员国采取一切适当措施,以确保不携带PPE或医疗设备的医疗设备。根据本建议书第8段可以在欧盟市场上投放的CE标记仅提供给医护人员,
通过了此建议:
1.为了确保在COVID-19爆发中提供个人防护设备和医疗设备以提供适当的保护,委员会邀请整个供应链中的所有经济运营商以及指定机构和市场监督机构在其部署中采取所有措施处置以支持旨在确保整个欧盟市场的个人防护设备和医疗设备的供应与不断增长的需求相匹配的工作。但是,此类措施不应对整体健康和安全水平产生不利影响,所有相关利益相关方应确保投放到欧盟市场的任何个人防护设备或医疗设备继续为用户提供足够的保护水平' 健康和安全。
合格评定程序
2、根据(EU)2016/425号条例的指定机构,应优先考虑并迅速进行PPE经济运营商在保护COVID-19时应保护的PPE经济运营商所有新提交的要求中的合格评定活动。
3、对于遵循非统一标准的技术解决方案生产的PPE产品,只要这些技术解决方案确保足够的保护水平,WHO关于适当选择PPE的建议可以用作此类技术解决方案的潜在参考来源。符合法规(EU)2016/425规定的适用基本健康与安全要求。
4、向遵循统一标准以外的其他技术解决方案生产的PPE产品颁发证书的认证机构,应立即将其颁发的证书和遵循的具体技术解决方案通知相关的通报机构以及根据(EU)2016/425条的其他认证机构。公告机构应通过根据法规(EU)2016/425第36条成立的公告机构小组的协调来交换此类信息。
5,就医疗器械而言,根据指令93/42 / EEC第11(13)条和法规(EU)2017/745的第59条,还应考虑成员国授权从合格评定程序中减损的可能性。后者在不需要指定机构介入的情况下也适用。
市场监督程序
6、成员国的相关市场监督机构应优先关注不合规的个人防护设备或医疗设备,这些设备会对其预期用户的健康和安全造成严重风险。
7、市场监督机构发现PPE或医疗设备可确保根据(EU)2016/425法规或93/42 / EEC指令或2017(EU)法规的基本要求确保足够的健康和安全水平/ 745,即使尚未按照协调规则完全完成包括CE标志在内的合格评定程序,它们仍可以授权在限定的时间内在欧盟市场上提供这些产品程序正在执行中。
8、不带CE标志的PPE或医疗设备也可以进行评估,并由相关成员国当局组织购买的一部分,前提是要确保此类产品仅在当前健康危机期间可供医护人员使用,并确保它们没有进入常规分销渠道,并可供其他用户使用。
9、市场监督机构应立即将其授予特定PPE或医疗设备的任何临时安排告知委员会和其他成员国。对于个人防护设备,应通过用于市场监视的信息和通信系统(ICSMS)来完成。
附:欧盟成员国名单(27国):
奥地利、比利时、保加利亚、塞浦路斯、捷克、克罗地亚、丹麦、爱沙尼亚、芬兰、法国、德国、希腊、匈牙利、爱尔兰、意大利、拉脱维亚、罗马尼亚、立陶宛、卢森堡、马耳他、荷兰、波兰、葡萄牙、斯洛伐克、斯洛文尼亚、西班牙、瑞典。
(来源:Mike外贸说)
以上内容属作者个人观点,不代表雨果网立场!本文经原作者授权转载,转载需经原作者授权同意。
































 闽公网安备35020602003453号
闽公网安备35020602003453号