






















































































 免费参与·100+跨境活动
免费参与·100+跨境活动 
 免费下载·4000+跨境资料
免费下载·4000+跨境资料 
 免费学习·2000+直播课程
免费学习·2000+直播课程 
 免费加入·15万+卖家交流群
免费加入·15万+卖家交流群 


2020-04-03 10:26

首先我们来关注一下最近闹得沸沸扬扬关于荷兰购买口罩质量问题事件。2020年4月2日,外交部发言人华春莹主持例行记者会,回应荷兰采购口罩“质量问题”。此外,商务部也披露了核实结果:为个人防护口罩,企业出口前已做说明。
问:近日,荷兰、比利时等部分欧洲国家媒体报道称,从中国购买的口罩不合格,存在质量问题。中方对此有何评论?
答:关于荷兰媒体称口罩存在质量问题,根据中方有关部门初步调查了解,那批口罩是荷兰代理商自己采购的, 中方企业发货前已告知荷方此批口罩为非医用口罩,出口报关手续也是以“非医用口罩”名义履行的。
在当前全球抗疫形势下, 中方急各国之所急,克服自身困难,有些企业加班加点,夜以继日,积极为国际社会提供各种防疫物资。我们一贯高度重视出口产品质量。
中方有关部门刚刚出台更加严格的监管措施,要求有关医疗物资出口企业在向海关报关时,必须提供书面或电子声明,承诺出口产品已取得我国医疗器械产品注册证书,符合进口国(地区)的质量标准要求。(出口新规详情查看:美国突然取消对中国KN95口罩标准认可!4月1日起医疗物资出口有新规!附CE/FDA认证资格认证指南)
口罩分为不同的防护标准和等级,也有日用防护和专业医用之分。当前,各国急需防疫物资,我们善意提醒使用方在购买和使用之前仔细核对产品用途和使用说明,以及是否符合采购方的使用标准,避免急中出错,误将非医用口罩配用于医用。
个别媒体在未弄清事实之前, 炒作所谓的中国产品质量问题,是不负责任、我希望他们不是别有用心的,因为这样做不利于国际抗疫合作。
提醒:海关已通知企业按照《商务部 海关总署 国家药品监督管理局公告2020年第5号 关于有序开展医疗物资出口的公告》要求,对出口医用口罩、医用防护服做出明确的质量监管措施,为落实好监管要求,海关要求各报关企业严格按照申报要求,在商品规格型号栏详细填报品牌、规格型号和用途,达到医用标准的必须填报医用。如发现商品实际属性为医用,填报为非医用情况,海关将按照不如实申报处理,请报关企业提醒“收发货人”落实好主体责任。
4月2日,商务部召开例行新闻发布会,商务部外贸司二级巡视员(副司级)刘长于就荷兰媒体报道从中国进口的口罩出现质量问题做出回应。
刘长于说,荷兰公司向中国相关企业采购的这批口罩为个人防护用的非医疗用口罩。有关企业出口时也做了说明:非医用口罩不能用于医疗用途,也不能用于在重症监护室工作的医护人员。
3月28日荷兰一些媒体报道称,荷兰从中国购买的60余万只口罩存在质量问题,被卫生部全部召回。
刘长于透露说,商务部注意到荷兰媒体相关报道,对此高度重视,第一时间向地方商务主管部门、相关出口企业进行多方核实。
刘长于说,“根据有关材料,我们了解到,荷兰公司向我相关企业采购的这批口罩为个人防护用的非医疗用口罩。有关企业出口时也做了说明。非医用口罩不能用于医疗用途,也不能用于在重症监护室工作的医护人员。”
刘长于表示,中国政府一贯高度重视医疗物资质量安全,对相关产品实行严格管理。在疫情防控的特殊时期,为进一步加强医疗物资出口质量监管,规范出口秩序,商务部会同海关总署、药监局发布公告,相关医疗产品出口必须取得我国医疗器械产品注册证书,符合进口国(地区)的质量标准要求。
刘长于也强调说:“我们希望国外采购方选择在我国药监部门注册的产品供应商,并在产品使用前进行相应的质量检验,严格按照产品适用范围和操作规程正确使用。如在采购和使用中出现有关问题,建议双方企业按商业化原则妥善协商解决。”
注意
近日,欧洲安全联盟(European Safety Federation)在其官方网站上发布文章表示,他们从不同渠道获悉“证书”用作PPE(包括FFP2/FFP3口罩)CE标记的依据,而这些“证书”没有法律价值,不能用作合格评定的结论。到目前为止,已经在CELAB、ICR Polska、ISET、ECM、NPS、CIC、Amtre Veritas和GTS的信头上看到了“证书”,目前尚不清楚这些文件是否真的是由上述组织签发的。
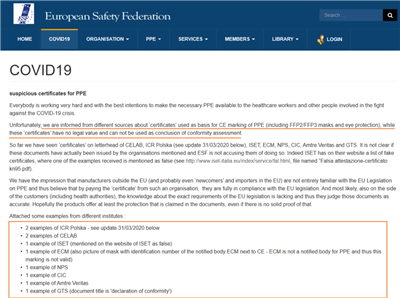
以下是来自不同机构的一些案例:
ICR Polska,2例
CELAB,2例
ISET,1例(已在ISET的网站上提到为假)
ECM,1例(口罩的CE标识旁边有公告机构ECM的识别号,但ECM不是个人防护用品的公告机构,因此该CE标识无效)
NPC,1例
CIC,1例
Amtre Veritas,1例
GTS,1例(文件标题是自我声明)
上述案例详情及具体图片,请查看欧洲安全联盟官网下的案例链接。
https://www.eu-esf.org/covid-19/4513-covid-19-suspicious-certificates-for-ppe
欧洲安全联盟表示,虽然目前当务之急是尽可能多地将口罩(和其他相关的个人防护用品)带入欧盟,以便保护医疗工作者。但不可接受的是不提供声称的、保护低于标准的口罩提供给了现在处于高风险的医护人员。根据(EU)2016/425号法规,防护面罩(如FFP2/FFP3)为III类PPE。这意味着合格评定包括:
1、由公告机构(Notified Body)进行的型式检验,合格后获发“EU型式检验证书”,简称Module B证书。
2、由公告机构进行的生产跟踪或随机抽查或体系审核,简称Module D证书或Module C2证书。
所以,目前只有获得欧盟 (EU) 2016/425 PPE法规口罩产品Module B、Module C2和/或Module D授权的认证公告机构才有权从事PPE个人防护口罩的CE认证。
鉴于疫情期间的PPE资源短缺及健康危机,欧盟委员会公布了关于合格评定和市场监督的(EU)2020/403号建议,允许成员国可以将个人防护用品投放到合格评定程序尚未完全完成的市场上。这仅适用于在危机期间由卫生保健主管部门购买的,但这并不意味着产品不必符合PPE法规中规定的适用基本健康和安全要求,进入正常销售链的个人防护用品仍然必须完全符合规定。
欧洲安全联盟提醒各相关利益方:检查您收到的个人防护用品“证书”是否正确命名为“EU type examination certificate (EU型式检验证书)”,以及它们是否由合格的公告机构颁发,公告机构的识别号必须包含在证书中。
在任何情况下,请查看“EU type examination certificate (EU型式检验证书)” 的措辞(或另一种欧盟语言中完全相同的措辞——请查看国家版本的立法,以获得正确的法律术语)。‘verification of compliance’, ‘certificate’, ‘certification report’等名称都不是正确的法律术语,因此具有此类名称的文件不是有效的EU型式检验证书。
如果上述“公告机构”的地址不在欧盟范围内,这已经强烈表明文件存在问题,因为个人防护用品的公告机构都设在欧盟成员国或有相互承认协议的一些国家。
还要查找公告机构的名称和编号(编号为4位)。为了确保公告机构确实是真实的且有相应资质的,您可以查看Nando数据库进行辨别。
欧洲安全联盟官网会定期更新PPE可疑证书的案例,详情可查看:https://www.eu-esf.org/covid-19/4513-covid-19-suspicious-certificates-for-ppe
出口欧盟CE认证或将改为MDR
新冠疫情爆发后,口罩作为防治疫情的必需品,从在特定领域中使用的用品,而一跃变为了现实中一罩难求的日用必需品。
中国作为世界工厂,一夜之间也蜂拥出现了几千家的口罩生产企业。这些口罩企业有相当多的瞄准了目前疫情严重的欧盟地区,希望将其的口罩投入欧盟市场,而欧盟市场的基本认证要求就是CE认证,它是打开并进入欧洲市场的“通行证”,是欧盟法律对限制类产品提出的强制性要求。
目前市场上新近完成的医用口罩CE证书基本上都是基于欧盟医疗器械指令MDD 93/42/EEC进行发放的。
然而这其中存在一个潜在的危机:市场上部分口罩的CE证书,可能还有1个多月就要换版了。
关于MDR (EU 2017/745)分析
2017年5月5日欧盟就发布了新版医疗器械法规MDR(EU 2017/745)。在2017年5月25日,MDR正式生效。老的医疗器械指令即MDD( 93/42/EEC)与新的MDR(EU 2017/745)指令的交替过渡期为三年。
也就是说从2020年5月26日, MDR指令在欧盟就将开始强制执行,它将完全取代过去老的医疗器械指令MDD (93/42/EEC)和老的有源植入医疗器械指令AIMDD(90/385/EEC)。
但是,对于已经在欧盟渠道正式上市的产品来说,老的MDD指令CE证书可以保持到2024年5月26日;
需要特别指出的是
☑ MDR强制执行后,新申请的CE认证必须按照MDR执行;
☑ 当前没有CE证书的产品,自5月27日起,必须按照MDR认证;
☑ 2020年5月26前签发的MDD证书,在有效期内仍然可以用,最晚到2024年5月26日;
☑ 原有MDD证书需在证书失效前换发 MDR。
具体欧盟CE的医疗指令过渡时间安排如下图所示:
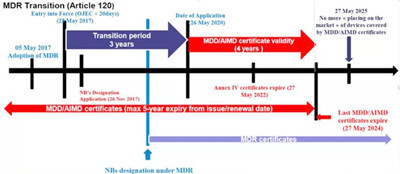
5月26日起施行的欧盟MDR指令,对目前医用口罩的CE认证,具体会有哪些影响?
1. 本次新冠疫情爆发期间所获CE认证的医用口罩,可以说95%以上的都是按照老的MDD指令进行的,可能要面临新版换证问题;
2. 根据目前欧盟最新统计,拥有老指令版本MDD(93/42/EEC)授权的NB公告机构共有56家,而符合MDR授权的NB公告机构目前则仅有12家而已。也就是说,从2020年5月26日开始,针对医用口罩的CE认证审核机构可选性降低了80%;
3. 由于欧盟MDR此类授权审核机构(NB:Notified Body)可选性的减少,必然造成医用口罩CE认证费用相当大的概率将有大幅提升的可能;
4. 新版MDR指令审核要求比老版MDD指令更为复杂,认证周期必然大幅度拉长,本次疫情期间有些机构声称的几天出证的可能性将基本为零;
5. 获得CE认证的口罩等医疗设备必须要求有指定的欧盟授权代表(简称"欧代"),这在以前MDD指令时对一些低风险产品其实有没有欧代监管并不严格,但在MDR指令后,即使是在一些电商平台上进行销售的医用产品也会要求提供必要的欧代信息;
6. MDR的体系审核流程和要求更为复杂与繁琐,举个例子如在MDR条款15中要求,医疗器械厂商应在其组织架构内,至少配备一名负责监管合规的人员,即合规负责人(Person responsible for regulatory compliance)。该人员应具备医疗器械领域的必要专业知识,并且有一系列的资格性证明要求(如法律、医学、药学、工程或其他相关学科,并且至少拥有一年与医疗器械法规事务或质量管理体系相关的专业经验);
7. 对于已经在欧盟渠道正式上市的产品来说,老的MDD指令CE证书可以保持到2024年5月26日。但如果企业产品在今年5月26日前,并未在欧盟市场销售的,原则上在今年5月26日后应该将老的MDD证书重新申请调整到MDR版本。
此次欧盟是直接发布的Regulation(法规),相比较之前的Directive(指令)其区别在于:提高了约束力,发布立即在欧盟成员国生效并成为有约束力的法律,此次的Regulation无需向Directive那样需要经过成员国转化成当地法律法规去落实实施。
因此,企业在申请医疗产品CE认证时,在过渡阶段请谨慎考虑是选用最新法规还是采用老的指令方案,同时也需要对NB机构的发证资格进行了解和确认以保证产品在欧盟市场销售的可延续性。
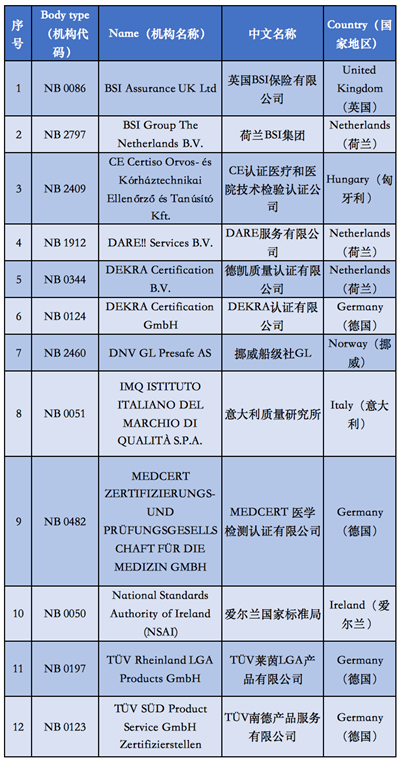
附①:2020年5月26日起
12家MDR授权机构清单
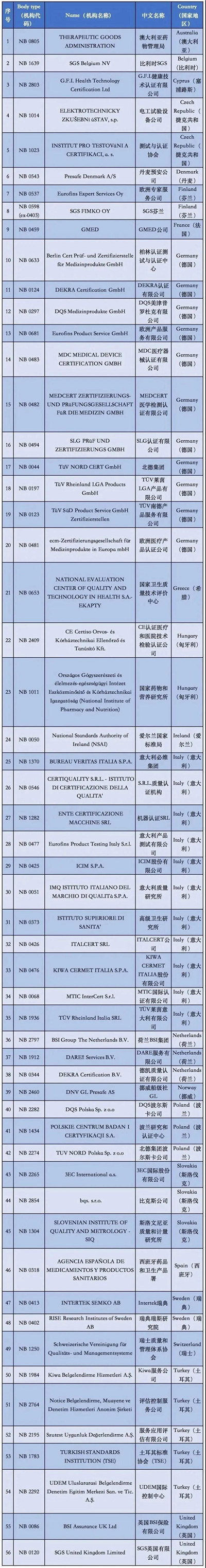
附②:2020年5月26日前
56家MDD授权机构清单
受疫情影响,很多国家和机构的政策都是频繁变动的,比如美国对于中国标准KN95口罩的态度,所以,MDR指令是否会实施,是否会延期实施,目前都还是未知数,只能静观其变!
欧盟CE认证资格指南
出口欧盟市场,CE认证必不可少。目前市场上出现了各色各样的医疗CE证书,让人眼花缭乱。在各种行业微信群里,经常可以看到有人发出一张所谓的CE证书,请大家帮忙辨别真伪。为了更好地帮助到大家,下面我们就来谈谈具体的鉴别方法。
查询CE证书的真伪,有多种方式,首先是最简单粗暴的一种。大的公告机构会在自己的官网上开放查询证书的窗口,当然,这种方式仅适用于发证机构正好提供了查询服务的情况。而对于未开放证书查询服务的机构,就不会奏效了。那么对于此类情况,当我们拿到一张医疗CE证书时,我们又该如何鉴别呢?
我们依然可以尝试从您手上这张证书的发放机构入手,去欧盟官网查询,看它是否具备欧盟医疗器械指令MDD 93/42/EEC或MDR医疗器械法规(EU) 2017/745及个人防护装备授权(EU)2016/425的相应认证资质。
欧盟官网MDD 93/42/EEC医疗器械指令授权的机构查询地址:
https://ec.europa.eu/growth/tools-databases/nando/index.cfm?fuseaction=directive.notifiedbody&dir_id=13
欧盟官网MDR (EU) 2017/745医疗器械法规授权的机构查询地址:
https://ec.europa.eu/growth/tools-databases/nando/index.cfm?fuseaction=directive.notifiedbody&dir_id=34
欧盟官网 (EU)2016/425个人防护装备授权的机构查询地址:
https://ec.europa.eu/growth/tools-databases/nando/index.cfm?fuseaction=directive.notifiedbody&dir_id=15550
通过欧盟官网可以看到,拥有MDD 93/42/EEC医疗器械指令授权的公告机构共有56家,具体的机构清单,公告号,以及其有资质审核的产品范围都详细罗列在上面。
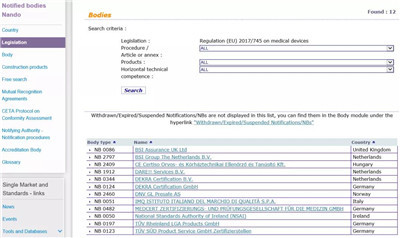
自2020年5月26日起,MDR (EU) 2017/745医疗器械法规将正式取代欧盟现行的MDD医疗器械指令强制实施,同样在欧盟官网可以查询到,拥有MDR授权的公告机构目前只有12家。
所以,如果您手上的医疗CE证书发证机构不在以上名单范围内,则说明它并不具备医疗产品的欧盟发证资质,更别谈CE证书的发放了,那么,很遗憾地说,您拿到的这张“”CE证书“是无效的。
另外,我们也可以从医疗器械产品CE认证的流程着手去分析,完成鉴别。
以口罩为例,首先,确认此口罩是否属于医疗器械。口罩分为医用口罩和防护口罩两种,如果是后者,则不属于医疗器械,不需要满足欧盟医疗法规要求,按照PPE个人防护指令完成CE认证即可。如果是医用口罩,则需要按医疗器械法规完成认证。而对于医用口罩来说,则需要进一步确认它是否无菌。如果是无菌医用口罩,在欧盟属于一类带灭菌医疗产品,必须按照医疗器械指令/法规MDD/MDR进行CE认证,这类情况是一定需要有授权的公告机构参与的。如果是非无菌医用口罩,则是按照医疗器械指令/法规MDD/MDR进行CE自我宣称,企业不需要通过公告机构认证,在准备好相应文件及测试报告等资料后,即可自行完成符合性声明。
就目前的情况而言,鉴于无菌医用口罩CE认证的难度较高且需要的时间较长,绝大部分的厂家都选择了非无菌医用口罩来生产和完成认证。
这里需要划个重点,既然是由制造商进行自我宣称CE符合性,又何来公告机构发放CE证书一说呢?如果不能发放CE证书,那很多企业所拿到的所谓证书又到底是什么呢?让我们找一些模板来看看:

请大家仔细研究一下其内容:“Verification of the presence of the Technical Files in regards of the Medical Devices Directive…” 意思是此证书证明了机构复核过该企业已经按照医疗器械法规要求来准备技术文档。
再来:“This document has been issued on voluntary basis and not as NB…” 意思是此证书是自愿性出具,不代表我以公告机构名义来执行了此事。
“…declares that the only scope of the assessment is to verify the existence of the declaration issued by the manufacturer or an applicant under its own responsibilities” 意思是此证书只是从他们的角度核实了制造商或申请人根据其本身的责任所发出的符合性声明是存在的。
因此,这类所谓的证书不是真正意义上由有授权的公告机构出具的CE证书。所以请大家务必擦亮眼睛鉴别真伪。
美国FDA认证应急授权
美国对于医疗器械的管理归口在美国食品药品监督管理局(FDA)的器械和辐照健康中心(CDRH)。医用口罩、体温计(包括耳温枪、额温枪、普通电子体温计和水银体温计)和在中等至高等风险下使用的防护服和隔离服都是二类医疗器械,需要申请510(k)。N95口罩虽然可以豁免510(k),但必须首先取得美国疾控中心NIOSH的N95证书。美国FDA对于所有的医疗器械仅有处方和非处方之分,并没有家用和专业医护人员使用的区别。
510(k)的申请流程如下:
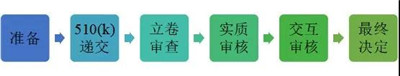
通常一个产品从启动510(k),准备测试和各类文件,直至最终的审核结束,需要历经8-10个月,而初次申请的企业,这个时间通常会更长。
除了510(k)以外,FDA要求所有的医疗器械企业都需要进行场所注册(Establishment Registration)和产品列示(Product Listing),这一要求对于应急使用授权的产品也不例外。对于所有的海外企业,在进行场所注册之前应先获得邓白氏编码,编码在中国由华夏邓白氏公司代理发放,免费的代码需要大约30天可以获得。获取邓白氏编码后,大约需要1-2周左右完成场所注册和产品列示。
无论是510(k),场所注册或者产品列示,FDA都不会向企业颁发任何证书,仅以美国FDA数据库中的数据为准。也就是说,大家见过的各种有着老鹰标记的证书都是没有任何效力的。
目前,新冠疫情在美国呈现明显爆发的趋势,各类医疗物资也趋于紧张。美国食品药品监督管理局(FDA)早在今年(2020年)2月初就开始对医疗器械潜在短缺的情况进行调查,为了应对各类医疗器械的紧缺FDA发布了各类应急使用授权(Emergency Use Authorization,EUA)。目前可以申请EUA的产品主要是未获上市的N95口罩、未获上市的新冠病毒诊断试剂、未获上市的酒精洗手液产品、已上市但需扩展用途的非侵入远程监护系统、已上市和未上市的呼吸设备。
新冠检测试剂:首当其冲的就是诊断试剂,FDA已经就该产品发布了第二版的指导原则。第一版指导原则主要针对美国国内临床实验室自我开发的检验方法的EUA申请,而第二版则纳入了针对生厂商的递交EUA申请的详细指导。EUA的申请要包含的内容与510(k)相似,需要提交检测试剂的描述、预期用途、性能评价报告、临床评价方案、稳定性试验方案、标签标识等。由于属于应急审批,FDA还要求企业必须提供针对患者和专业人员的明白纸(Fact Sheet)。目前海河咨询已经完成了新冠病毒胶检测试剂盒胶体金法的EUA申请。
口罩:其次紧缺的就是口罩。口罩在美国有多种类别,海河也专门针对口罩注册途径举办过专题线上研讨会,针对不同类别的上市途径进行过说明。FDA也针对口罩发布了2轮的EUA通知。
第一轮的EUA允许直接使用满足如下条件的口罩:
(1)根据NIOSH42 CFR part 84作为非动力空气净化过滤面罩口罩批准的所有一次性过滤式口罩呼吸器(包括N95口罩)。以及
(2)经NIOSH批准但已过制造商推荐的保存期限的过滤面罩口罩,供医护人员在医疗环境中使用,以防止医护人员由于面罩口罩短缺,而暴露于病原性生物空气传播颗粒中。
考虑到这样的措施还是无法保障美国市场的口罩供应,FDA又在近期发布口罩的EUA申请,特别地,没有获得NIOSH批准的口罩也可以进行EUA的申请,但是必须符合:
条件1-满足特定国家/地区性能标准,包括
澳大利亚:P2,P3
巴西:PFF2,PFF3
欧盟:FFP2,FFP3
日本:DS/DL3,DS/DL2
韩国:Special 1st
墨西哥:N100, P100, R100, N99, P99, R99, N95, P95, R95
和/或
条件2-获得特定国家上市许可,包括:
欧盟:CE认证;
澳大利亚:ARTG注册
加拿大:注册证;
日本:医疗器械注册证。
非侵入远程监护系统:例如可穿戴的设备,手持设备或者家用固定监护设备。产品类别包括体温计、心电图机、心电软件、血氧仪、血压计、呼吸监测设备等。这些监护设备应当具有潜在的网络连接能力,包括蓝牙,Wi-Fi或蜂窝网络等,并且能将检测数据直接传输给医疗机构。为了应对新冠疫情,对于已经获得FDA许可上市的非侵入远程监护系统,在应急使用期间,FDA将允许其在适应症,宣称功能,硬件或软件等各方面的有限的变更,而不需要对变更递交510(k)申请。
呼吸设备:适用于应急使用授权的呼吸类设备指向具有呼吸衰竭或呼吸不足情况的病人提供通气和呼吸支持的设备,包括但不限于医院用连续呼吸机,家用连续呼吸机,应急使用呼吸机等。FDA仍然建议医疗机构使用FDA许可的传统/全功能的呼吸机,但是考虑到尽可能多的可获得性,对于已经获得FDA许可上市的呼吸设备,在应急使用期间,FDA将允许其在适应症,宣称功能,硬件或软件等各方面的有限变更,而不需要对变更递交510(k)申请。此外,对于呼吸设备,FDA还开通了EUA申请。对于有兴趣进行申请的生产商,FDA将提前介入与生产商沟通相关申请文件的要求以及对相关的申请进行加急审核。
酒精洗手液产品:之前酒精洗手液产品在美国是按照非处方类药品进行管理的,洗手液中的酒精则是按照API进行管理的。目前,FDA已经发布通知,不会对提供用于洗手液的酒精的生产商或者洗手液的生产商采取行动,即可以认为,FDA将允许用于洗手液的酒精和基于酒精的洗手液在满足限定条件下直接在美国销售。海河咨询特别提醒:对于洗手液的生产商仅限定适用于药剂师以及联邦设施,不适用于其他商业生产商。
(来源:海运信息)
以上内容属作者个人观点,不代表雨果网立场!如有侵权,请联系我们。




























 闽公网安备35020602003453号
闽公网安备35020602003453号